Using protein interaction database and support vector machine to improve gene signatures for prediction of breast cancer recurrence
Abstract
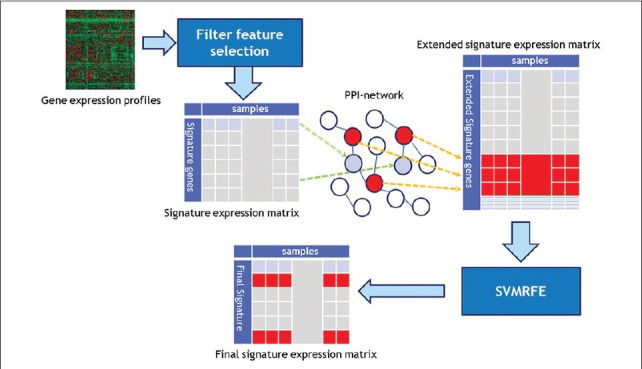
Numerous studies used microarray gene expression data to extract metastasis-driving gene signatures for the prediction of breast cancer relapse. However, the accuracy and generality of the previously introduced biomarkers are not acceptable for reliable usage in independent datasets. This inadequacy is attributed to ignoring gene interactions by simple feature selection methods, due to their computational burden. In this study, an integrated approach with low computational cost was proposed for identifying a more predictive gene signature, for prediction of breast cancer recurrence. First, a small set of genes was primarily selected as signature by an appropriate filter feature selection (FFS) method. Then, a binary sub-class of protein-protein interaction (PPI) network was used to expand the primary set by adding adjacent proteins of each gene signature from the PPI-network. Subsequently, the support vector machine-based recursive feature elimination (SVMRFE) method was applied to the expression level of all the genes in the expanded set. Finally, the genes with the
Journal of Medical Signals and Sensors
Wolters Kluwer -- Medknow Publications
Using Protein Interaction Database and Support Vector Machines to Improve Gene Signatures for Prediction of Breast Cancer Recurrence
Mohammad Reza Sehhati, Alireza Mehri Dehnavi, [...], and Shaghayegh Haghjoo Javanmard
Abstract
Numerous studies used microarray gene expression data to extract metastasis-driving gene signatures for the prediction of breast cancer relapse. However, the accuracy and generality of the previously introduced biomarkers are not acceptable for reliable usage in independent datasets. This inadequacy is attributed to ignoring gene interactions by simple feature selection methods, due to their computational burden. In this study, an integrated approach with low computational cost was proposed for identifying a more predictive gene signature, for prediction of breast cancer recurrence. First, a small set of genes was primarily selected as signature by an appropriate filter feature selection (FFS) method. Then, a binary sub-class of protein-protein interaction (PPI) network was used to expand the primary set by adding adjacent proteins of each gene signature from the PPI-network. Subsequently, the support vector machine-based recursive feature elimination (SVMRFE) method was applied to the expression level of all the genes in the expanded set. Finally, the genes with the highest score by SVMRFE were selected as the new biomarkers. Accuracy of the final selected biomarkers was evaluated to classify four datasets on breast cancer patients, including 800 cases, into two cohorts of poor and good prognosis. The results of the five-fold cross validation test, using the support vector machine as a classifier, showed more than 13% improvement in the average accuracy, after modifying the primary selected signatures. Moreover, the method used in this study showed a lower computational cost compared to the other PPI-based methods. The proposed method demonstrated more robust and accurate biomarkers using the PPI network, at a low computational cost. This approach could be used as a supplementary procedure in microarray studies after applying various gene selection methods.



