An in silico Post-GWAS Analysis of C-Reactive Protein Loci Suggests an Important Role for Interferons
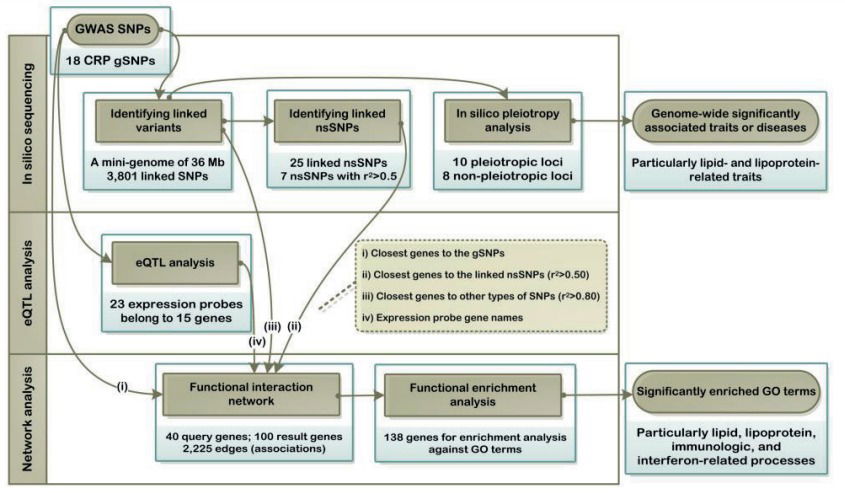
BACKGROUND: Genome-wide association studies (GWASs) have successfully identified several single nucleotide polymorphisms (SNPs) associated with serum levels of C-reactive protein (CRP). An important limitation of GWASs is that the identified variants merely flag the nearby genomic region and do not necessarily provide a direct link to the biological mechanisms underlying their corresponding phenotype. Here we apply a bioinformatics-based approach to uncover the functional characteristics of the 18 SNPs that had previously been associated with CRP at a genome-wide significant level.
METHODS AND RESULTS: In the first phase of in silico sequencing, we explore the vicinity of GWAS SNPs to identify all linked variants. In the second phase of expression quantitative trait loci analysis, we attempt to identify all nearby genes whose expression levels are associated with the corresponding GWAS SNPs. These 2 phases generate several relevant genes that serve as input to the next phase of functional network analysis. Our in silico sequencing analysis using 1000 Genomes Project data identified 7 nonsynonymous SNPs, which are in moderate to high linkage disequilibrium (r(2)>0.5) with the GWAS SNPs. Our expression quantitative trait loci analysis, which was based on one of the largest single data sets of genome-wide expression probes (n>5000) identified 23 significantly associated expression probes belonging to 15 genes (false discovery rate <0.01). The final phase of functional network analysis revealed 93 significantly enriched biological processes (false discovery rate <0.01).



